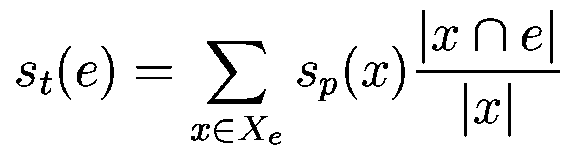| |
sgp-2 is a program to predict genes by comparing anonymous genomic
sequences from different species. It combines tblastx, a sequence
similarity search program, with geneid, an "ab initio" gene prediction
program (http://www.sanger.ac.uk/Software/formats/GFF/). In "assymetric" mode, genes are predicted in one sequence
from one species (the target sequence), using a set of sequences
(maybe only one) from the other species (the reference
set). Essentially, geneid is used to predict all potential exons along
the target sequence. Scores of exons are computed as
log-likelihood ratios, function of the splice sites defining the exon,
and of the coding bias in composition of the exon sequence as measured
by a Markov Model of order five.
From the set
of predicted exons, geneid assembles the gene structure (eventually
multiple genes in both strands) maximizing the sum of the scores of
the assembled exons, using a dynamic programming chaining algorithm.
When using a reference set of sequences,
ideally we would like to incorporate into the scores of the
candidate exons, the score of the
optimal alignment at the amino acid level between the target exon
sequence and the counterpart homologous sequene in the reference
set. If a substitution matrix, for instance from the blosum
family, is used to score the aligment, the resulting score can also be
assumed to be a log-likelihood ratio: informally, the ratio between
the likelihood of the aligment when the amino acid sequences code for
proteins functionally related, and the likelihood of the alignment,
otherwise. In principle, thus, this score could be naturally added to the geneid score for the exon. tblastx provides an appropiate shortcut to find
often a good enough approximation to
such an optimal aligment, and infer the corresponding score:
the optimal alignment can be assumed to correspond to the
maximal scoring HSP overlapping the exon.
However, when dealing with the fragmentary nature of the mouse
shotgun genomic sequence, often different regions of a candidate exon
sequence will align optimally to different mouse shotgun
sequences. Thus, in the approach used here, we identify the optimal
HSPs covering each fraction of the exon, and compute
separately the contribution of each HSP into the score of the
exon. In the next section, we describe in detail how this computation
is performed.
|
Scoring of candidate exons
|
Let 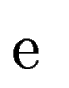 be one of the candidate exons predicted by geneid along the a
query DNA sequence be one of the candidate exons predicted by geneid along the a
query DNA sequence 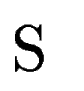 . In sgp-2 the final score of , . In sgp-2 the final score of , 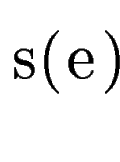 , is
computed as , is
computed as
where 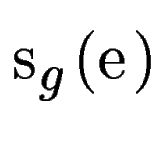 is the score
given by geneid to the exon , and is the score
given by geneid to the exon , and 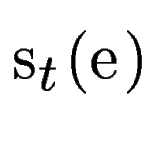 is the score derived from
the HSPs found by a tblastx search overlapping the exon . Both
scores are log-likelihood ratios (and we compute both base
two). Assuming independence between their distribution, they could be
summed up into a single likelihood ratio. However, the assumption of
independence is not realistic, depends on the probability of
the sequence of , assuming that codes for a protein, while
depends on the probability of the optimal aligment of
with a sequence fragment of the mouse genome, assuming that both
sequences code for related proteins. Obviously, these two
probabilities are not independent. Their joint distribution could only
be investigated-at least empirically-if the Markov Model of coding
DNA used in geneid, and the substituion matrix used by tblastx were
inferred from the very same set of coding sequences. Since this is not
the case, we use an ad-hoc coefficient, is the score derived from
the HSPs found by a tblastx search overlapping the exon . Both
scores are log-likelihood ratios (and we compute both base
two). Assuming independence between their distribution, they could be
summed up into a single likelihood ratio. However, the assumption of
independence is not realistic, depends on the probability of
the sequence of , assuming that codes for a protein, while
depends on the probability of the optimal aligment of
with a sequence fragment of the mouse genome, assuming that both
sequences code for related proteins. Obviously, these two
probabilities are not independent. Their joint distribution could only
be investigated-at least empirically-if the Markov Model of coding
DNA used in geneid, and the substituion matrix used by tblastx were
inferred from the very same set of coding sequences. Since this is not
the case, we use an ad-hoc coefficient, 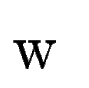 , to weight the
contribution of tblastx search, into the final exon
score.
We compute in the following way.
Let , to weight the
contribution of tblastx search, into the final exon
score.
We compute in the following way.
Let
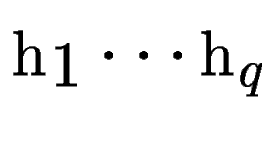 be the set of HSPs found by tblastx after
comparing the query sequence against a database of DNA sequences
(figure be the set of HSPs found by tblastx after
comparing the query sequence against a database of DNA sequences
(figure ![[*]](cross_ref_motif.gif) , A).
First, we find the maximum scoring projection of the HSPs onto
the query sequence. We simply register the maximum score
among the scores of all HSPs covering each
position, and then partition the query sequence in equally maximally
scoring segments , A).
First, we find the maximum scoring projection of the HSPs onto
the query sequence. We simply register the maximum score
among the scores of all HSPs covering each
position, and then partition the query sequence in equally maximally
scoring segments
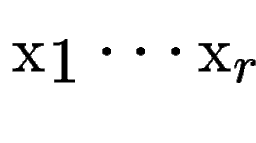 , with scores , with scores
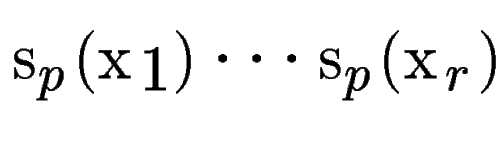 (figure , B).
Then, for each predicted exon (figure ,C), we find (figure , B).
Then, for each predicted exon (figure ,C), we find  the set of maximally scoring segments overlapping
the set of maximally scoring segments overlapping
where 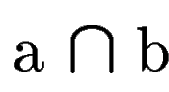 denotes the overlap betwen sequence segments denotes the overlap betwen sequence segments 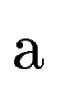 and and 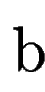 , and , and
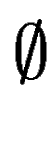 means no overlap. We compute in the following way: means no overlap. We compute in the following way:
where 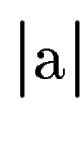 denotes the length of sequence segment .
That is, each exon gets the score of the maximally scoring HSPs
along the exon sequence proportional to the fraction of the HSP
covering the exon. In other words, is the integral of the
maximum scoring projection function within the exon interval.
Once the scores denotes the length of sequence segment .
That is, each exon gets the score of the maximally scoring HSPs
along the exon sequence proportional to the fraction of the HSP
covering the exon. In other words, is the integral of the
maximum scoring projection function within the exon interval.
Once the scores 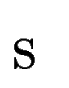 have been computed for all predicted exons in the
sequence , gene prediction proceeds as usual in geneid: the gene
structure is assembled maximizing the sum of scores of the assembled
exons. have been computed for all predicted exons in the
sequence , gene prediction proceeds as usual in geneid: the gene
structure is assembled maximizing the sum of scores of the assembled
exons.
In the practice, we run sgp-2 in the following way. Given a DNA query
sequence (here, human chromosome sequences), and a database of DNA
sequences (here, shotgun mouse genomic sequences), we run the query
sequence against the databse using tblastx. Results of tblastx search are parsed to obtain the
maximum scoring projection of the HSPs onto the query sequence
using a simple perl script. The parsing includes discarding all HSPs
below a given bit score cutoff, substrating this value from the score
of the remaining HSPs, and weighting the resulting score by (see
above).
The maximum scoring projection is given to geneid in GFF
format `General Feature Format' (Durbin and Haussler, http://www.sanger.ac.uk/Software/formats/GFF/). geneid uses it to rescore the exons predicted along the query sequence as
explained, and assembles the corresponding optimal gene
structure.
Figure:
Rescoring of the exons predicted by geneid according with the results of a tblastx search
(To enlarge the figure, click right mouse button and select "View Image" from panel).
![\includegraphics[bb= 87 120 502 718,clip,angle=270]{algo1.ps}](img20.gif) |
|
|