| geneid documentation: | 7. Using sequence homology information |
| Table of contents: |
| HSPs: |
|
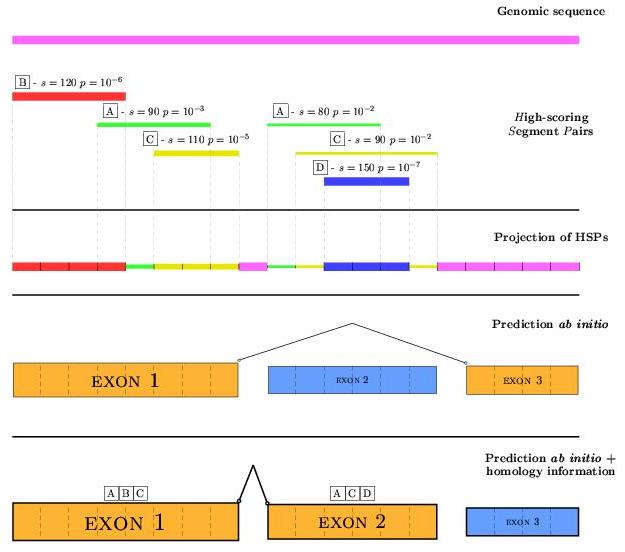
Given an blast aligment, a set of High-score Segment Pairs (HSPs)
are produced to represent parts common in both sequences. Given the
interesting sequence (input to geneid), numerous pairwise
alignments can be done by using sequences related to the original by
many causes such as containing an homologous gene (syntenic gene prediction).
geneid will increase proportionally the score of predicted exons
which overlap the HSPs obtained by previous pairwise aligments. To sum up
the information coming from several aligments, a projection of HSPs over the
sequence is performed, taking always the highest score among the HSPs
being in every position of the input sequence. Exon positions overlapping
regions without homology support will be given a value NO_SCORE
(see chapter 8. geneid
parameter file)
|
| Frame definition in blast and geneid |
|
It is important to understand that frame definition in blast and
geneid is different and therefore a simple translation from one into
another format is needed. Altough this conversion is implemented inside
geneid, it might be necessary to keep it in mind to analyze and
understand the new results.
In blast, there are 3 frames (1,2 or 3)
corresponding to the mathematical modulus operation computed following this
formula:
i.e. Given a sequence which its first position is called 1, one HSP which starts
at the position 7 is said to be in frame 1 (7 modulus 3 = 1), while a HSP
which starts at the position 9 is said to be in frame 3 (9 modulus 3 = 0
and 0 is coded by 3).
IMPORTANT: HSPs might appear in the negative (reverse) strand of the
input sequence. In that case, the HSPs coordinates must be translated into
coordinates regarding to the reverse reading direction before computing the
frame.
In geneid, the frame is defined as the number of nucleotides (0,1,2)
from the first nucleotide in the exon sequence to the the first nucleotide
in the first COMPLETE codon translated from the genomic sequence
(see section Frame and Remainder (chapter 2)).
Obviously, it is necessary a method to convert geneid frames (exons)
into blast frames (HSPs) to use this information properly. The
following formula makes both types of frame definitions compatible
Given an exon and a HSP (both in the same strand):
blastFrame(HSP) = (starting(exon) + geneidFrame(exon)) % 3; notice: blastFrame = [1,2,3] and geneidFrame = [0,1,2]. |
Enrique Blanco Garcia © 2003