Roderic Guigó, IMIM and UB, Barcelona
COMPUTATIONAL GENE IDENTIFICATION
NOTE: click on the images through this document
to download higher quality postscript images.
The Problem
The Gene Identification Problem can be formulated as the problem of
deducing the aminoacid sequences encoded in a given DNA genomic sequence.
Why is the problem relevant?
-
From an applied standpoint, it is of great relevance given the Genomic
Projects underway. Automatic generation of very long anonymous DNA genomic
sequences it is already a fact (visit, for the instance, the
Sanger Center ftp site,
where raw sequence is being produced at a rate of
100 Kb/day, as of march 1997).
Reliable computational analysis may substantially
decrease the cost and time required to characterize such data.
-
From a basic standpoint, the problem is also very relevant. It is the problem
of understanding the way genes are specified in the genome. Being able
to deduce the aminoacid sequences encoded in a given DNA genomic sequence
by reying only on the sequence
is, after all, what it means deciphering the genetic code.
Why is the problem difficult?
In higher eukariotc organims, genes are neither contiguos nor continuous.
First, genes coding for different proteins are separated by
large intergenic regions that do not code for proteins.
Second, a given protein
sequence is not usually specified by a continuous DNA sequence, but
genes are often splitted in a number (maybe large) of (small) coding
fragments known as Exons, separated by (larger) non-coding intervining
fragments known as Introns (See figure below).
Often, intronic and intergenic DNA makes
most of the genome in high eukariotic organisms. In the human genome,
for instance, only a very small fraction of the DNA, which can be as low
as 2%, corresponds to protein coding exons.
In the next sections, we show that although signals exist on the DNA
sequence that instruct the cellular machinery along the pathway from DNA
to protein sequences, our knowledge of the way such signals are recognized
and processed by the cell is very limited, and it is usually impossible
to infer the genes encoded in a given DNA sequence by reliying only on these
signals
A few types of signals on the DNA sequence
are involved in gene specification
The figure below schematizes the pathway from DNA to protein sequences in
a higher eukariotic cell. The main steps in this pahtway are:
-
Transcription.
The continuous sequence of DNA corresponding to a single gene is
copied to an RNA sequence.
-
Splicing.
The primary RNA transcript is spliced to remove intron sequences, producing
a shorter RNA molecule, known as messenger RNA (mRNA).
-
Tranlation.
The mRNA sequence is translated into protein sequence by a
sub-cellular structure known as ribosome. The ribosome binds to an initiation
codon, and scans
the sequence synthesizing the amino acid sequence specified by
consecutive non-overlapping codons. Scanning of the mRNA proceeds until
the ribosome finds one of the three codons not specifying amino acids (the
Stop Codons).
At that point, elongation of the amino acid sequence ends, and
the final protein product is released.
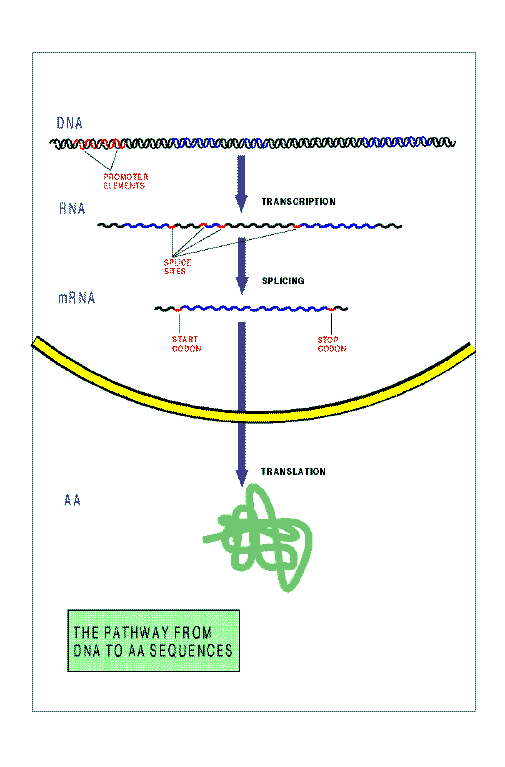
Signals exist in the DNA sequence---short strings of nucleotides---, which
instruct the cellular machinery during these steps.
The Promoter Elements, and the
Transcription Termination Motif
during transcription,
the Donor Sites and Acceptor Sites
during Splicing, and the Initiation Codon and the
Stop Codon during Translation.
Although eventually recognized by
the cellular machinery through intermediate
RNA molecules, the signals involved in gene specification are all
ultimately encoded in the primary DNA sequence.
DNA signals involved in gene specification are
aparently ill-defined and highly unspecific
DNA signals involved in gene specification are ill-defined,
they lack generality, and are highly unspecific;
with currently available detection methods, it is usually
impossible to distinguish the signals truly processed by the cellular
machinery from those---much more frequent---apparently non functional. As a
consequence, attempting to predict gene structure by processing solely DNA
sequence signals
often results in a computationally untreatable combinatorial explosion
of potential products.
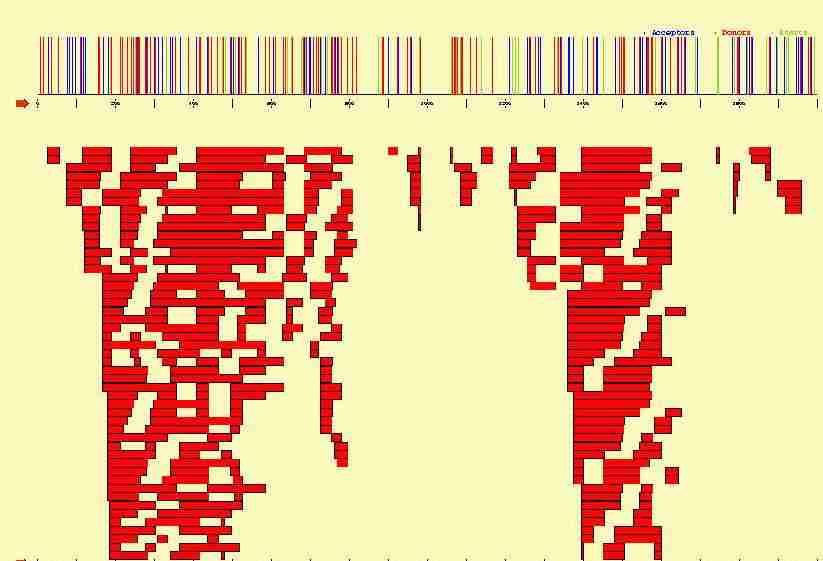
In the figure below, we plot the potential start sites, acceptor and donor
sites that can be identified along the 2000 bp long sequence containing
the three exon beta-globin gene. Sites have been identified using
a Position Weigth Matrix, with a cutoff such that no potential true sites
are missed. From such signals, hundreds of potential exons can be
constructed, which in turn can be combined into milions of potential genes.
The cell apparently finds precissely its way through this puzzle,
and only one (or a few) of such genes appear to be actually specified.
Information other than sequence signals can be
used to infer the genes potentially encoded in a DNA sequence.
Information from a number of sources, other than the sequence signals
recognized by the cellular machinery, can be used to infer the genes encoded
by the cellular machinery. Roughly, this information can be categorized as
follows:
- Intrinsec. Information derived only from the query
sequence itself (without reference to other known sequences).
- Signal. Information derived from sequence signals.
Sequence signals can be not only identified, but also scored.
A wide variety of methods exist to score and locate sequence signals,
collectively known as Search by Signal methods.
- Content. Information derived from the fact that coding
regions in the DNA exhibit peculiar sequence statistical properties. A wide
variety of coding measures (or statistics) have been developed over the years.
They are collectively known as Search by Content methods.
- Extrinsec. Information derived by comparing
the query sequence with other known sequences (in the public databases).
Usually, coding regions such as amino acid sequences or ESTs, but also
regulatory sequence such as promoters, or even intrinsically non coding
sequences, such as repeats.
In the figure below, we plot how this additional information can help us
to localize the exons of the beta-globin gene.
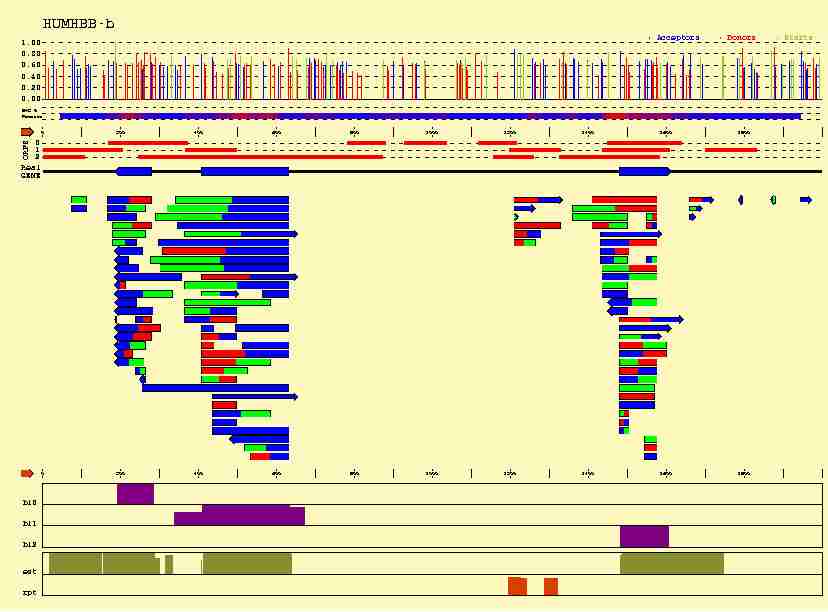
-
Sequence signals
(Acceptor sites in blue,
Donor sites in red, and Initiation Codons in green) can not only be identified,
but also scored; Although functional sequence patterns show sequence
conservation, this conservation is usually not absolute. Among the possible
patterns, some of them are more likely than others, and this "likelihood"
can be somehow measured.
- A number
of coding sequence statistics can be computed along the sequence. Here,
we have computed a measure of the 3-base periodicity on an sliding window
along the sequnce. Coding regions are known to be characterized by an strong
3-base periodicty. Thus, highly periodic regions (higher in red along the line
plotted below the predicted sites) are more likely to be coding.
All this intrinsec information can be used to score the predicted exons,
and eventually filter out unlikely candidates.
-
The position of Stop Codons can be used to delimitate long ORFs where exons
can occur, and to establish the frame in which a predicted exon can be read.
Only frame compatible exons (exons matching adjacent colors) can be assembled
into functional genes, limiting substantially the number of possible candidates.
- Database matches are extremly useful, since they constitute very
strong evidence of the existence of the genes---but they not always exist.
In this case,
matches to amino acid sequences in the SWISSPROT database in the three frames
have been ploted in purple, matches to cDNAs sequences in the dbEST database
in green, and matches to repetitive sequences in the REPEAT
database in orange.
Up to date Electronic Biobliographies on Computational Gene Identification
are maintained by
Roderic
Guigo (i Serra), IMIM and UB. rguigo@indy.imim.es