| Comparative Gene Prediction | ||||||
|
|
||||||
|
Comparative genomics is the analysis and comparison of genomes from
different species. The purpose is to gain a better understanding of
how species have evolved and to determine the function of genes and
non-coding regions of the genome.
Researchers have learned a great deal about the function of human
genes by examining their counterparts in simpler model organisms such
as the mouse. Genome researchers look at many different features when
comparing genomes: sequence similarity, gene location, the length and
number of coding regions within genes, the amount of
non-coding DNA in each genome, and highly conserved regions maintained
in organisms as simple as bacteria and as complex as humans.
On the other hand, finding similarities is not as much important as
finding differences. The comparative approach also points out those
features which are unique for a given phylogenetic group or
particularly a species. Species specific functions can be involved in,
for instance, pathogenicity, resistance to antibiotics, and so on, but
also will result on more complex phenotypic characters such as the
human ability to speak. | ||||||
| Overview | ||||||
|
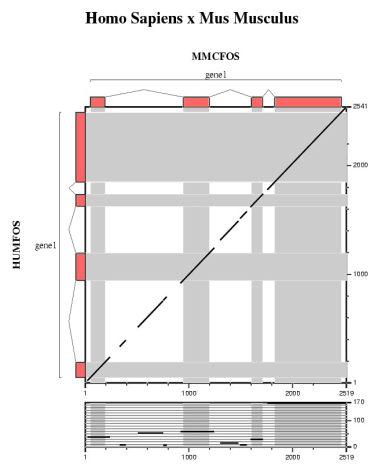
In this section we are going to run several ab initio gene prediction programs on a particular genomic DNA sequence. Thus, we can compare which elements are absent or present in all the predictions. Then, a comparative gene prediction program will be used to take advantage of the homology between the same gene in two species: human and mouse.
The programs we are going to use are geneid, genscan
and fgenesh. After that, blast will be used to compare
human and mouse sequences. Finally, we will run sgp2
(syntenic gene prediction tool) to build the prediction
taking into account the homology between both genomes.
| ||||||
|
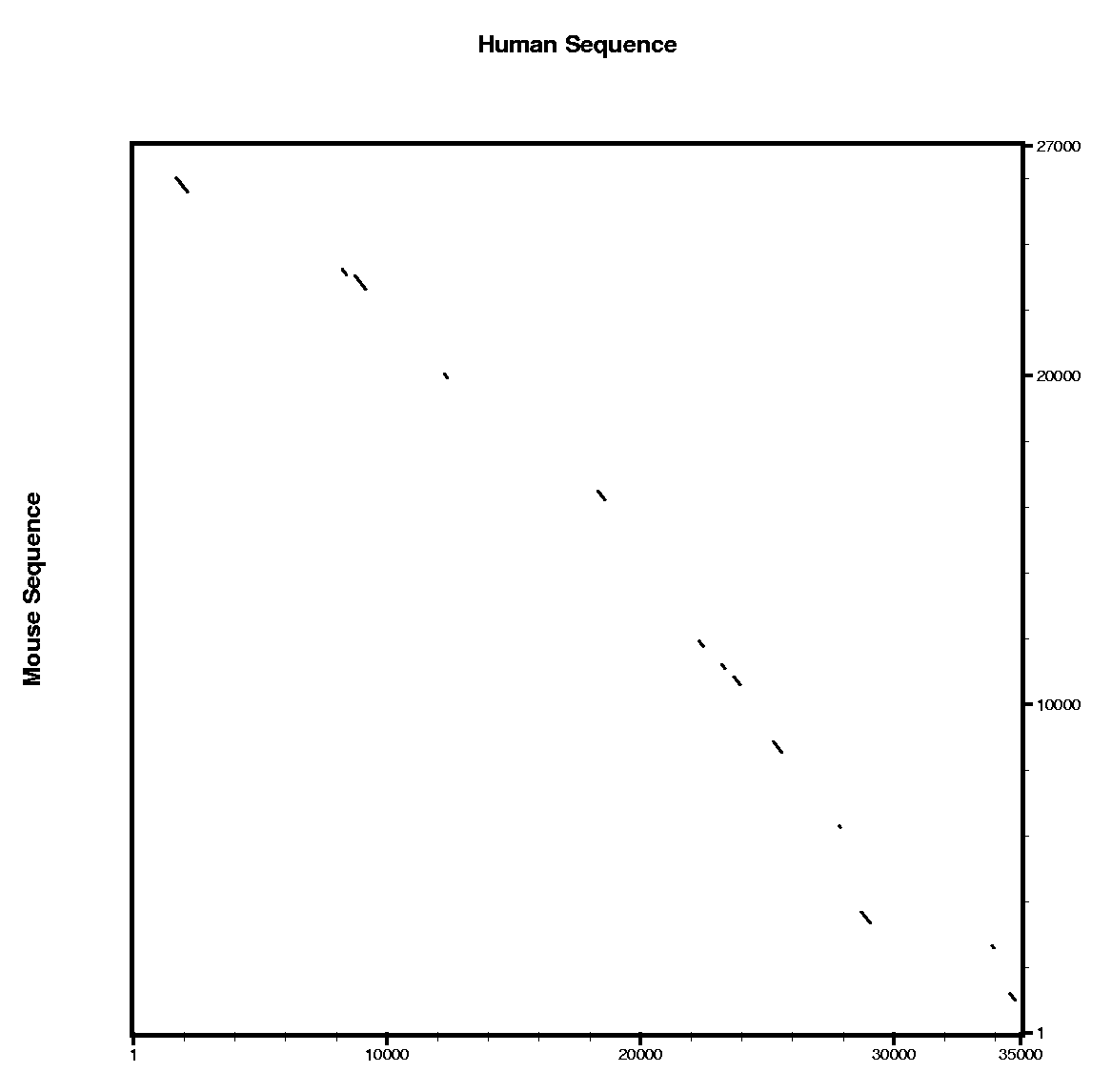
We are going to work with this
Human sequence, which is stored in FASTA format. We also provide
the homologous region in the mouse genome in this
Mouse sequence.
| ||||||
| Ab initio gene finding | ||||||
|
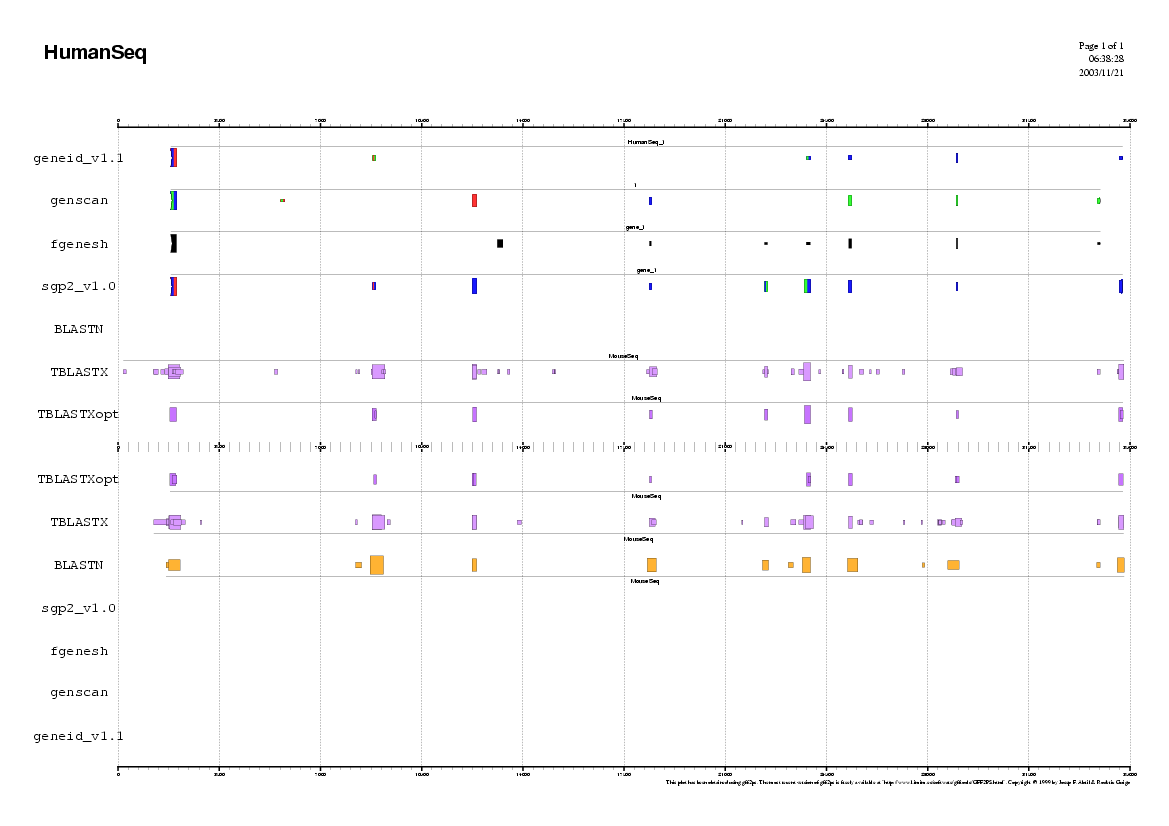
In the first approach, we will use all the ab initio tools
from the Gene Prediction section and compare the result of the three
programs. You could open a simple word processor and paste the results
of each gene-finding program in order to compare the coordinates of the
predicted exons.
Step 1
Analyzing the Human sequence.
In order to use geneid follow these steps:
| ||||||
|
In order to use genscan follow these steps:
| ||||||
|
In order to use fgenesh follow these steps:
Some questions:
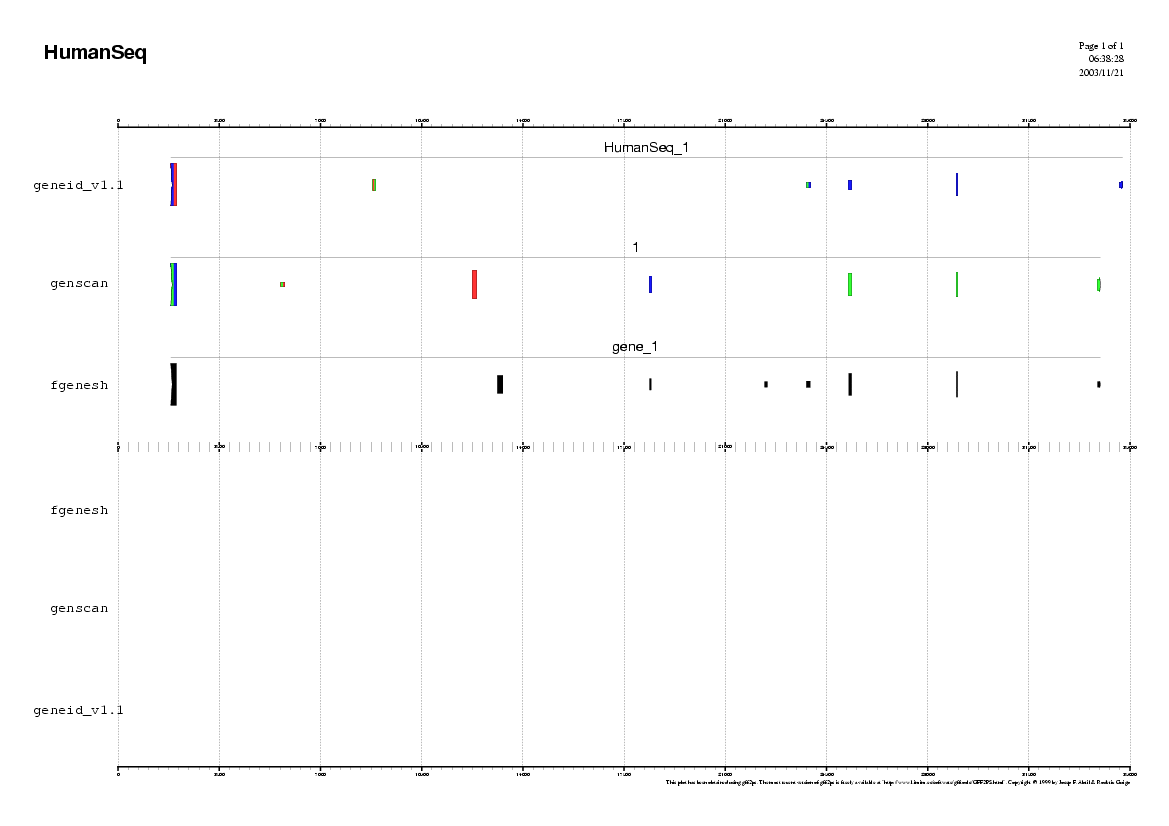
Step 2
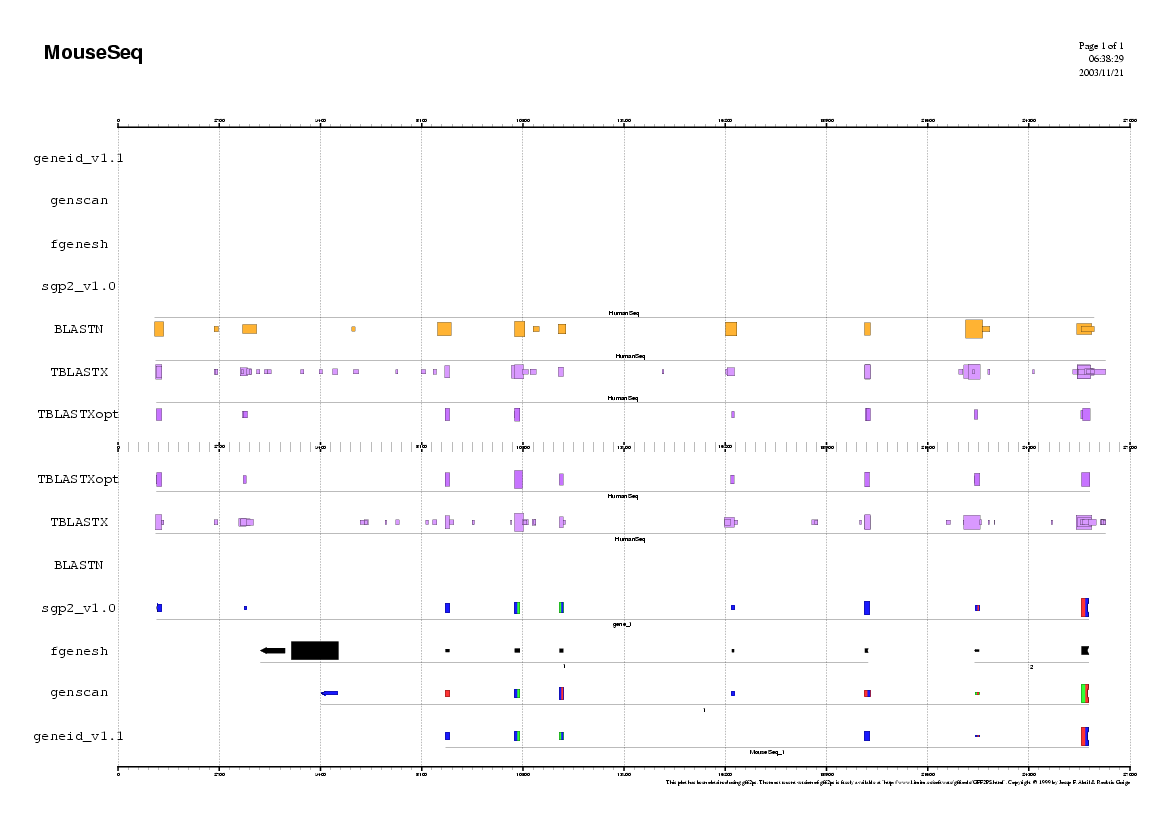
Analyzing the Mouse sequence
(Repeat the same procedure as in human)
Some questions:
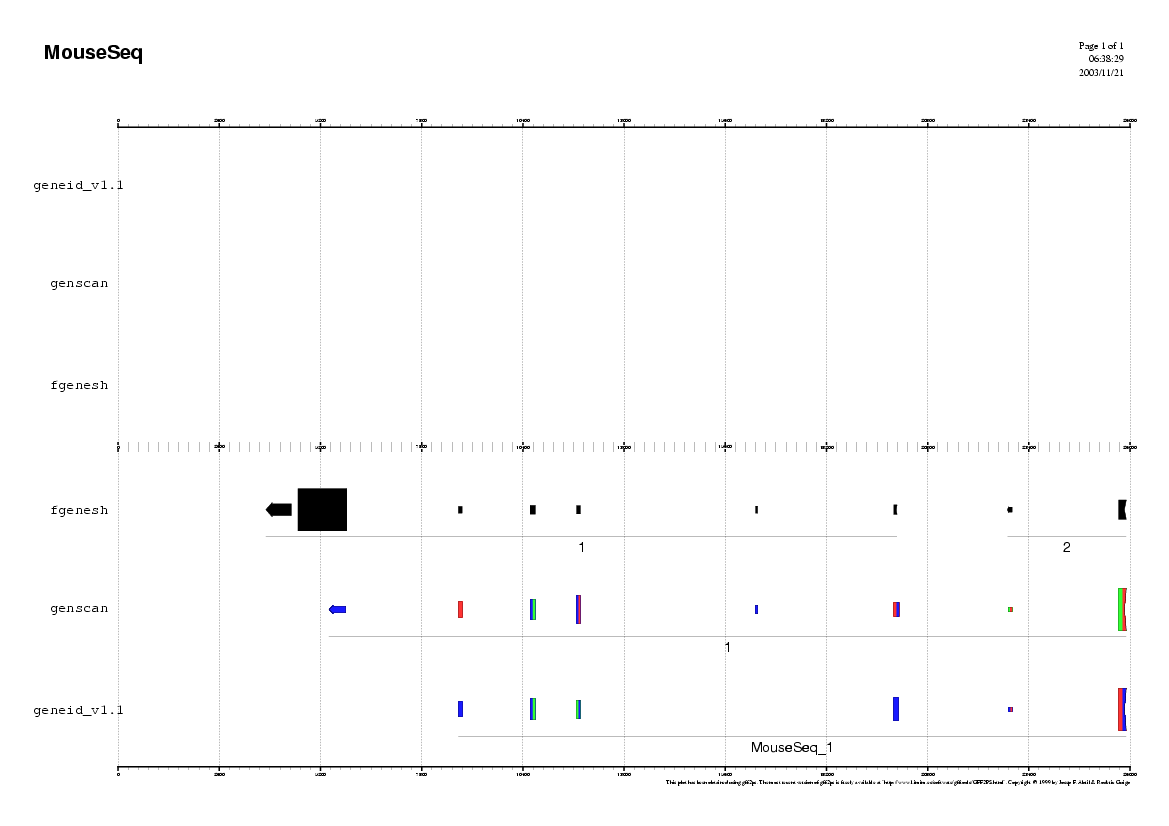
Do you observe any common pattern between human and mouse predictions ?
| ||||||
| Comparing human and mouse sequences | ||||||
|
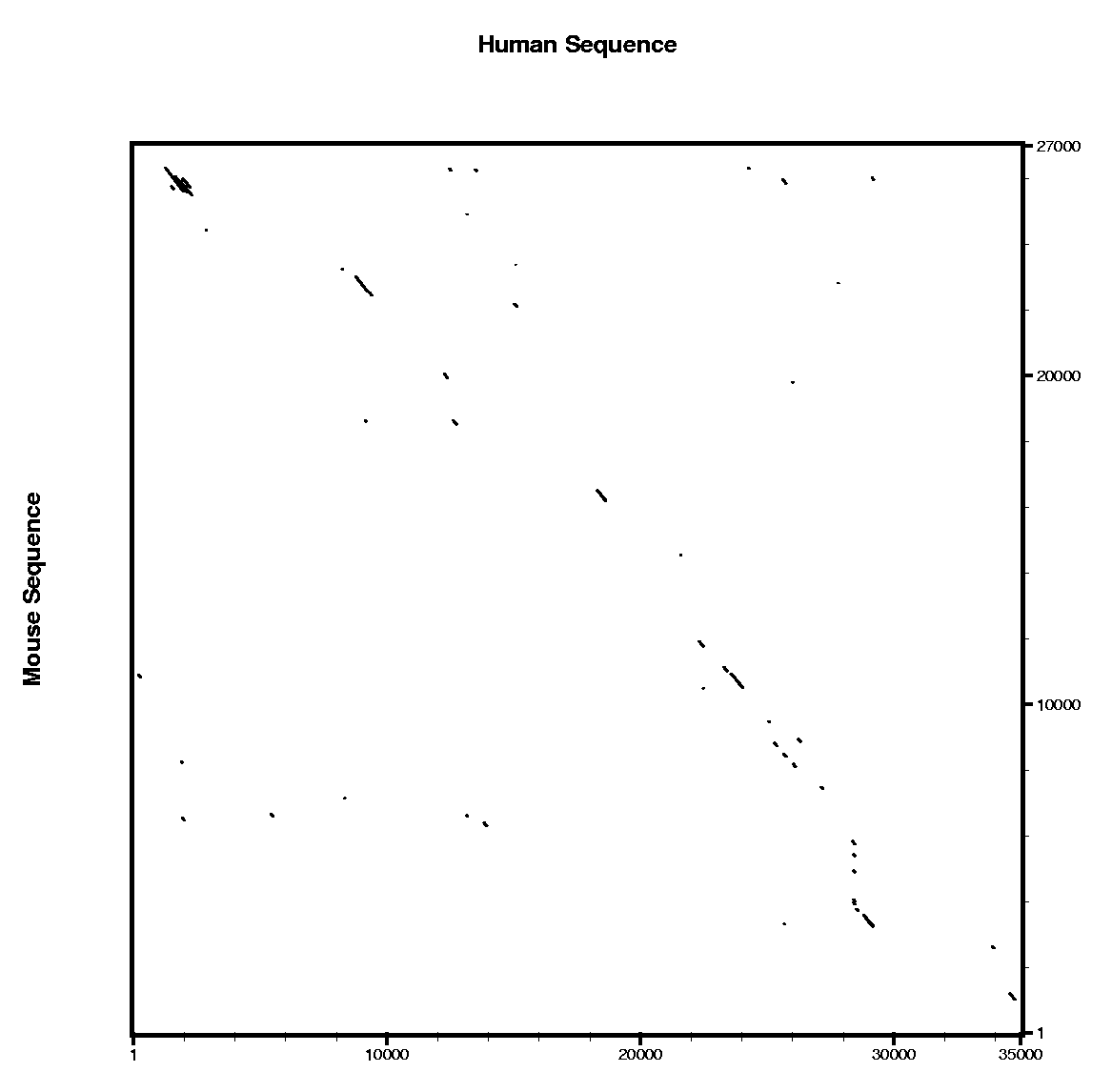
In order to use blastn follow these steps:
In order to use tblastx follow these steps:
Are the predicted exons supported by conserved regions ?
Other programs to align and visualize pairs of large genomic sequences are: gff2aplot, Vista and Pipmaker. | ||||||
| Using comparative gene finding tools | ||||||
|
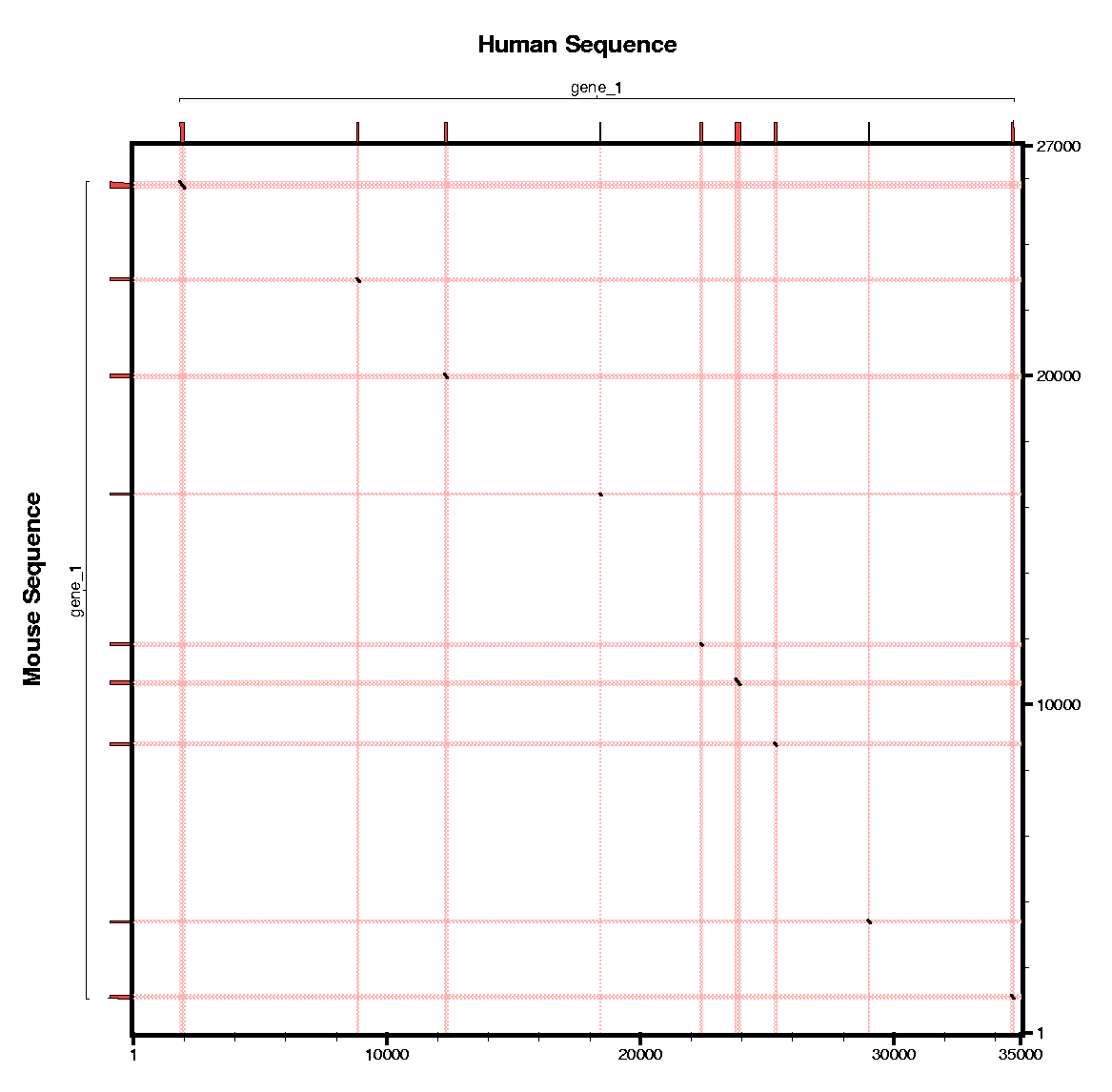
In this section we will use sgp2 to make the predictions
using the conservation pattern between human and mouse.
Some questions:
There are other programs that use genomic comparison to improve gene prediction: twinscan and slam. | ||||||
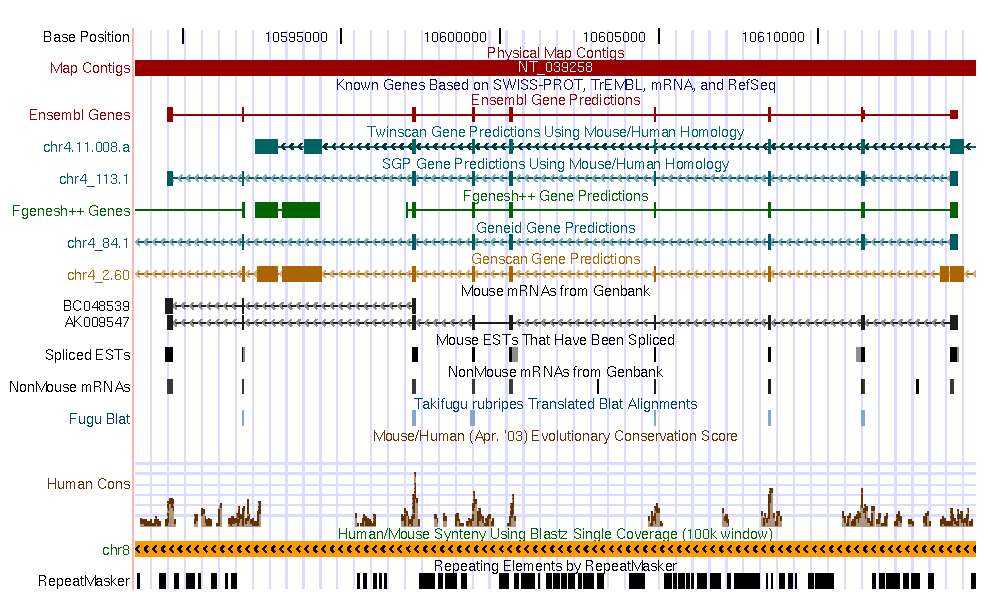
| Current annotations in the genomic DNA sequence | ||||||
|
Go to the UCSC genome browser , and look for the annotation of this region in the human genome. Open another window and look for the annotation of the mouse sequence in the mouse genome annotation.
Some questions:
The predictions that you can see are consistent with UCSC genome browser annotations
in both genomes?
|
||||||