Unequal usage of codons in the coding regions appears to be a universal feature of the
genomes across the phylogenetic spectra. This bias obeys mainly to (i) the uneven usage of
the amino acids in the existing proteins and (ii) the uneven usage of synonymous codons. The
bias in the usage of the synonymous codons correlates with the abundance of the corresponding
tRNAs. The correlation is particularly strong for highly expressed genes. Codon usage is
specific of the taxonomic group, and there exist correlation between taxonomic divergence and
similarity of codon usage.
By comparing the frequency of codons in a region of an species genome read in a given frame
with the typical frequency of codons in the species genes, it is possible to estimate a
likelihood of the region coding for a protein in such a frame. Regions in which codons are
used with frequencies similar to the typical species codon frequencies are likely to code
for genes (exons) while regions which codons distribution is uniform -- 1/64 (0.015625) --
could be considered as non-coding regions (introns).
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Let S a sequence of DNA, the higher the number of codons in S matching the more frequent codons in the table, the higher the coding potential of the sequence S (the probability to be encoding a protein). This value can be computed as:
where
This expression is known as likelihood ratio and it is usually computed
in logarithmics terms to reduce the magnitude of the numbers (log-likelihood
ratio).
1. Scan a sequence, computing the protein coding potential:
Here, you can find the human codon usage table
in a more manageable format (tabular format).
| We are going to write a program in order to measure the coding potential of a sequence in Fasta format taking into account a given table of codon usage. |
#!/usr/bin/perl -w
use strict;
# if the number of input arguments is lower than 2
# return a message showing the error
if (scalar(@ARGV) < 2) {
print "dnaloglkh.pl codontable DNAsequence\n";
exit(1);
}
# create two vars contaning the filenames
my $filecodontable = $ARGV[0];
my $filesequence = $ARGV[1];
# open the first file: table of codon usage frequencies
if (!open(PROPCODONS,"< $filecodontable")) {
print "dnaloglkh.pl: the file $filecodontable can not be opened\n";
exit(1);
}
# load the frequencies of codon usage into the hash table %pcodons
# this hash is indexed by a triplet of nucleotides or codon
my %pcodons;
my ($codon,$freq);
my $line;
# for each line in the file (tableofcodons) do
while ($line = <PROPCODONS>) {
# extract the two values or columns with a regular expression
# A group of letters and a decimal number
($codon,$freq) = ($line =~ m/(\w+) (\d+\.\d+)/);
# load the hash table with the frequency of the current codon
********* FILL IN 1 *********
}
close(PROPCODONS);
This sketch of program verifies that the user has provided 2 arguments in the input. The first one is the name of the file containing the table of codon usage. Once, the file has been opened, the content of the file (pairs codon,probability) will be save into a hash table called %pcodons. Please, complete the program with a single instruction to load the current probablity into the hash indexed with the current codon.
##########################################################
# open the DNA sequence (second file)
if (!open(SEQUENCE,"< $filesequence")) {
print "dnaloglkh.pl: the file $filesequence can not be opened\n";
exit(1);
}
# FASTA format:
# >header containing a description of the sequence
# AGACTGACTTAC
# AGACTGACTTAC
# AGACTGAC
my $iden = <SEQUENCE>; # reading the header of the sequence
my @seql = <SEQUENCE>; # load the complete sequence into an array
my $seq = ""; # use a var to save the whole sequence inside
close(SEQUENCE);
# concat every segment (line) in the array into the var $seq
my $i=0;
while ($i < scalar(@seql)) {
chomp($seql[$i]);
$seq = ********* FILL IN 2 *********
$i = $i + 1;
}
$seq = "\U$seq";
# we are going to use the array @codons to store all of the codons in
# the input DNA sequence. To split the sequence in segments of length
# three, a regular expression will be used searching for all of the
# possible groups of three symbols (option g)
my @codons = ($seq =~ m/.../g);
# the likelihood ratio is for a current triplet, the ratio between
# the probability computed from the table of codon frequencies divided
# by the random probability (uniform), that is, 1/64. After this,
# the log must be applied to obtain the log-likelihood ratio.
# It is recommended the use of logs to manage calculations involving
# small numbers between 0 and 1. Log of the product is equal to the
# the sum of logs and the log of the division is the substraction of logs
my $r = 0.0;
$i=0;
while ($i < scalar(@codons)) {
********* FILL IN 3 *********
$i = $i + 1;
}
printf "%.2f\n",$r;
If you can not suceed, you can take a look at the complete
program, here.
2. Compute the protein coding potential in the three reading frames:
Unfortunately, as you have seen the log-likelihood value computed for both cases is very similar so that we can not distinguish among coding and non coding regions. As you know, a DNA sequence can be translated into a series of amino acids using three different reading frames and we are probably using the wrong frame right now.
| We will modify the previous program to read an extra argument defining the reading frame (0,1,2) that will be used to measure the coding potential in the input sequence. |
Let us suppose that there is third argument (the reading frame) that will be
saved into the variable $ARGV[2]. Just before converting the sequence
into a list of codons (before the line my @codons = ($seq =~ m/.../g);),
we can delete zero, one or two characters from the beginning of the sequence with the
command substr().
$seq = substr($seq,$ARGV[2]);The function substr(s,p,l) extracts a subsequence of "s", starting from the point "p" that contains "l" symbols. If the third parameter is omitted, then the substring will be constructed from "p" to the end of the sequence "s".
$ ./dnaloglkh.pl propcodons.txt exon.fa 0
3. Compute the protein coding potential using sliding windows in the three reading frames at the same time:
In practice, the problem is not to determine the likelihood that a given sequence is coding
or not, but to locate the (usually small) coding regions within large genomic sequences. The
typical procedure is to compute the value of a coding statistic in successive (overlapped)
sliding windows, and record the value of the statistic for each of the windows. This generates
a profile along the sequence in which peaks may point to the coding regions and valleys to
the non-coding ones.
#!/usr/bin/perl -w
use strict;
# if the number of input arguments is lower than 3
# return a message showing the error
if (scalar(@ARGV) < 3) {
print "dnaloglkh.pl codontable DNAsequence frame\n";
exit(1);
}
# Load the arguments into three vars
my $filecodontable = $ARGV[0];
my $filesequence = $ARGV[1];
my $frame = $ARGV[2];
# open the first file: table of codon usage frequencies
if (!open(PROPCODONS,"< $filecodontable")) {
print "dnaloglkh.pl: the file $filecodontable can not be opened\n";
exit(1);
}
# load the frequencies of codon usage into the hash table %pcodons
# this hash is indexed by a triplet of nucleotides or codon
my %pcodons;
my ($codon,$freq);
my $line;
# for each line in the file (tableofcodons) do
while ($line = <PROPCODONS>) {
# extract the two values or columns with a regular expression
# A group of letters and a decimal number
($codon,$freq) = ($line =~ m/(\w+) (\d+\.\d+)/);
# load the hash table
$pcodons{$codon}=$freq;
}
close(PROPCODONS);
##########################################################
# open the DNA sequence (second file)
if (!open(SEQUENCE,"< $filesequence")) {
print "dnaloglkh.pl: the file $filesequence can not be opened\n";
exit(1);
}
# FASTA format:
# >header containing a description of the sequence
# AGACTGACTTAC
# AGACTGACTTAC
# AGACTGAC
my $iden = <SEQUENCE>; # reading the header of the sequence
my @seql = <SEQUENCE>; # load the complete sequence into an array
my $seq = ""; # use a var to save the whole sequence inside
close(SEQUENCE);
# concat every segment (line) in the array into the var $seq
my $i=0;
while ($i < scalar(@seql)) {
chomp($seql[$i]);
$seq = $seq . $seql[$i];
$i = $i + 1;
}
$seq = "\U$seq";
# using the function to define the operation of log-likelihood
printf "%.2f\n",compute_loglkh($seq,$ARGV[2]);
##############################################################
##############################################################
############# FUNCTIONS ####################
##############################################################
##############################################################
# NAME: compute_loglkh
# GOAL: compute the log-likelihood of a sequence using
# given reading frame
# PARAMS: first - DNA sequence
# second - reading frame = {0,1,2}
# RETURNS: the log-likelihood ratio (coding vs non-coding region)
sub compute_loglkh {
my $seq = $_[0];
my $frame = $_[1];
# the function substr(s,p,l) extract a subsequence of "s" from
# the position "p" having "l" symbols (if omitted, extract from that
# point to the end
$seq = substr($seq,$frame);
# we are going to use the array @codons to store all of the codons in
# the input DNA sequence. To split the sequence in segments of length
# three, a regular expression will be used searching for all of the
# possible groups of three symbols (option g)
my @codons = ($seq =~ m/.../g);
# the likelihood ratio is for a current triplet, the ratio between
# the probability computed from the table of codon frequencies divided
# by the random probability (uniform), that is, 1/64. After this,
# the log must be applied to obtain the log-likelihood ratio.
# It is recommended the use of logs to manage calculations involving
# small numbers between 0 and 1. Log of the product is equal to the
# the sum of logs and the log of the division is the substraction of logs
my $r = 0.0;
my $i=0;
while ($i < scalar(@codons)) {
$r = $r + log($pcodons{$codons[$i]}) - log(1/64);
$i = $i + 1;
}
return $r;
}
| We will add some commands to compute the coding potential in the three reading frames on series of windows or consecutive segments of DNA. |
In order to find a coding region (exon) in a long sequence, we should compute
the likelihood ratio in separate windows of a given length. Once the value
has been obtained for the three frames, the window must be shifted a set of
fixed positions (overlap).
#!/usr/bin/perl -w
use strict;
# if the number of input arguments is lower than 4
# return a message showing the error
if (scalar(@ARGV) < 4) {
print "dnaloglkh.pl codontable DNAsequence Lwindow Loverlap\n";
exit(1);
}
# Load the arguments into four vars
my $filecodontable = $ARGV[0];
my $filesequence = $ARGV[1];
# Length and overlap of the window to scan the sequence
my $lengthwindow = $ARGV[2];
my $overlap = $ARGV[3];
# Scan the three possible frames for every window
$i = 0;
while ($i <= length($seq) - $lengthwindow) {
# extract the segment of sequence corresponding to that window
# the function substr(s,p,l) extract a subsequence of "s" from
# the position "p" having "l" symbols
my $seqwindow = substr($seq,** FILL IN 4 **,** FILL IN 5 **);
# compute the log-likelihood ratio for every reading frame
my $frame0 = ********* FILL IN 6 *********
my $frame1 = ********* FILL IN 7 *********
my $frame2 = ********* FILL IN 8 *********
# take the maximum value. Display the value and the position
my $maximum = max($frame0,$frame1,$frame2);
printf "%d %.5f\n",$i,$maximum;
# increase $i with the $overlap value
$i = $i + $overlap;
}
# NAME: max
# GOAL: compute the maximum value in a list of N values
# PARAMS: a list of values
# RETURNS: the maximum value in the list
sub max {
my $i = 1;
my $max = $_[0];
while ($i < scalar(@_)) {
if ($_[$i] > $max) {
$max = $_[$i];
}
$i = $i + 1;
}
return $max;
}
$ ./dnaloglkh3.pl propcodons.txt hs.fa 120 10 > cu.datThe file cu.dat contains the log-likelihood ratio computed for every 120 bps-window, shifting from 10 to 10 bps in the sequence HS, according to the table of human codon usage.
To analyze this information, we can visualize the results in a graphical way using the program gnuplot:
$ gnuplot
gnuplot> plot 'cu.dat' with impulsesto obtain a plot like this:
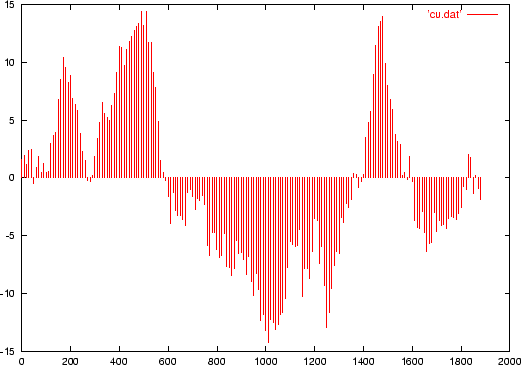 |
187 - 278 409 - 631 1482 - 1610
If you have any problem, download the complete program dnaloglkh3.pl.